






Machine Learning Yields More Efficient Hydrogen Combustion Reactivity Modeling
February 15, 2024
By Kathy Kincade
Contact: cscomms@lbl.gov
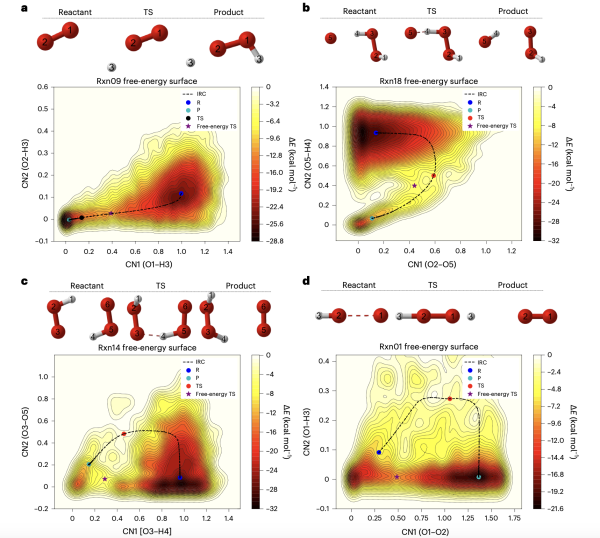
Free-energy surface reconstructed from metadynamics using a hybrid model for hydrogen combustion. Reactant, transition state, and product geometry are shown above the free-energy surface, with oxygen in red and hydrogen in silver and the atomic index labeled.
Science Breakthrough
Researchers from Lawrence Berkeley National Laboratory (Berkeley Lab), University of California Berkeley, and Penn State University demonstrated that active learning-based machine learning (ML) atomic force prediction offers a more efficient and computationally affordable alternative to ab initio molecular dynamics (AIMD) in hydrogen combustion modeling.
They trained a physics-inspired ML neural network called NewtonNet to predict atomic forces and potential energy surface (PES) used in modeling chemical reactivity. With this new hybrid model, the researchers realized two orders of magnitude reduction in computational cost when simulating hydrogen combustion reaction, enhancing the prediction of free-energy change in the transition-state mechanism for hydrogen combustion reaction channels.
This advance could help boost the development and adoption of hydrogen as an alternative clean energy fuel source for commercial and industrial applications.
Science Background
Understanding the details of a chemical reaction – a process that leads to the chemical transformation of one set of chemical substances to another – is a critical component of hydrogen combustion modeling. However, chemical reactions are difficult to study because they are rare events in which molecules transition from one low-energy configuration to another by crossing an energy barrier. Identifying such an event using standard AIMD simulation is time-consuming and computationally demanding for two reasons:
- The configuration associated with the energy barrier is not thermodynamically favorable, so barrier crossing may not occur even after tens of thousands AIMD steps.
- Each AIMD step involves computing the atomic forces that determine the motion for each atom. Forces are determined by performing an electronic structure calculation that takes quantum mechanical effects into account; the cost of this computation scales cubically with respect to the number of atoms.
To address the first challenge, metadynamics – a computer simulation method common in computational physics, chemistry, and biology in which a bias potential defined in terms of a small set of collective variables (CVs) is used to describe the low-dimensional reaction coordinates – is applied in the force calculation to accelerate barrier crossing and enhance configuration sampling. Metadynamics has the benefit that at finite temperatures one can determine the free energy transition state that is more informative than just the PES for chemical reactivity.
To overcome the second challenge, a neural network such as NewtonNet is used to predict atomic forces without performing expensive electronic structure calculations. However, to obtain accurate forces, the neural network needs to be trained with properly selected data; training with configurations only in the stable region of the PES is likely to produce inaccurate atomic forces for configurations that are near the barrier crossing region (the desired transition state). Furthermore, machine learning models must be trained on even more unintuitive regions of data – for example, to learn not to visit unphysical or inaccessible energy states.
Toward this end, active learning – a type of supervised learning in which an algorithm can choose the data it wants to learn from – offers a more effective training approach because new configurations generated from metadynamics can augment the training data and retrain the neural network when the accuracy of its prediction becomes less reliable. But at some point the cost of retraining an ML model from each round of active learning data becomes unsustainable for complex potential energy surfaces, as we see in hydrogen combustion. In this case, less reliable ML steps are directly replaced with the expensive ab initio calculation; but because this happens relatively infrequently, the overall greater efficiency of the ML model still dominates the longer accessible timescales reached by the hybrid model.
Science Breakdown
The success of this approach depends largely on which CVs are used to perform the metadynamics, and the selection of good CVs is not trivial. Collective variables are typically found using “chemical intuition” – unwritten guidelines applied by researchers to find the right synthesis conditions for various compounds. Our new ML/physics-inspired method uses diffusion mapping (an automated dimension-reduction and feature-extraction approach) to identify and assess the quality of the CVs. We have found that CVs that are highly correlated with diffusion coordinates are suitable for use with metadynamics to identify chemical reactions.
While applying ML to predict the atomic forces does not eliminate the need for quantum mechanical calculations in chemical reaction studies, using active learning to collect the data used to train the neural network is a more efficient way to apply electronic structure calculations and ensure accurate force prediction. Furthermore, a hybrid approach using AI and physics allows us to gain the benefits of longer simulation timescales (at least two orders of magnitude) while maintaining good chemical accuracy.
This work has its roots in an ongoing U.S. Department of Energy (DOE) Scientific Discovery Through Advanced Computing (SciDAC) chemical sciences partnership that is studying chemical reactions. A hydrogen combustion dataset developed at Berkeley Lab served as the benchmark dataset for studying the chemical reactions; simulations were run at the National Energy Research Scientific Computing Center (NERSC), a DOE user facility located at Berkeley Lab.
Research Leads
Teresa Head-Gordon: Kenneth S. Pitzer Theory Center and Department of Chemistry, UC Berkeley; Chemical Sciences Division, Berkeley Lab
Chao Yang: Applied Mathematics & Computational Research Division, Berkeley Lab
Co-authors
Xingyi Guan, Joseph P. Heindel: Kenneth S. Pitzer Theory Center and Department of Chemistry, UC Berkeley; Chemical Sciences Division, Berkeley Lab
Taehee Ko: Penn State University
Publications
Using machine learning to go beyond potential energy surface benchmarking for chemical reactivity, Nat Comput Sci 3, 2023, 965–974.
Using Diffusion Maps to Analyze Reaction Dynamics for a Hydrogen Combustion Benchmark Dataset, Chem. Theory Comput, 2023, 19, 17, 5872-5885.
A benchmark dataset for Hydrogen Combustion, Scientific Data 9, 2022, Article number 215.
Funding
CPIMS program, Office of Science, Office of Basic Energy Sciences, Chemical Sciences Division of the U.S. Department of Energy for support of the machine learning approach to hydrogen combustion. U.S. Department of Energy via the SciDAC program for the collective variables.
User Facilities
NERSC
About Berkeley Lab
Founded in 1931 on the belief that the biggest scientific challenges are best addressed by teams, Lawrence Berkeley National Laboratory and its scientists have been recognized with 16 Nobel Prizes. Today, Berkeley Lab researchers develop sustainable energy and environmental solutions, create useful new materials, advance the frontiers of computing, and probe the mysteries of life, matter, and the universe. Scientists from around the world rely on the Lab’s facilities for their own discovery science. Berkeley Lab is a multiprogram national laboratory, managed by the University of California for the U.S. Department of Energy’s Office of Science.
DOE’s Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit energy.gov/science.